Torsten H. Struck
Frontiers in Evolutionary Zoology
A major problem in phylogenetic or phylogenomic studies is that they might be hampered by signal stemming from other process than the descent-with-modification process. Therefore, we are developing new methods to ameliorate the impact of such processes in phylogenetic reconstructions.
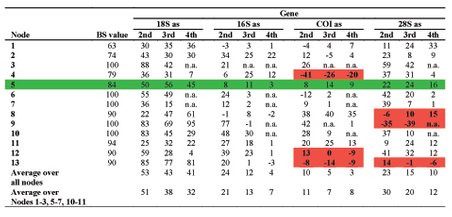
Processes such as lateral gene transfer, incomplete lineage sorting, compositional biases, mutational saturation or increased substitution rates, but also wrong assignment of a paralog to a ortholog group, so that the gene tree is inferred instead of the species, introduce artificial signal into the phylogenetic reconstruction and thus conceal the phylogenetic signal present in the dataset. Therefore, we develop new methods to determine the incongruence between different data (e.g., genes, but also morphological data) as well as some of their possible sources such as saturation, compositional biases or increased substitution rates more precisely. One such method is the PABA approach (see to the right), which assesses the congruence between partitions on a node-by-node manner rather than an all or nothing approach.
For these different approaches, we developed the program TreSpEx, which exploits the information present in the tree reconstruction of single gene analyses of phylogenomic datasets to detect potential paralogs or contaminations in the dataset, conduct PABA analyses in an automatic manner as well as to determine long branch and saturation indices for each taxon within the individual partition of a phylogenomic dataset. Implementations in the program are automatic BLAST searches of affected potentially paralogous sequences against selected NCBI databases to assess if a paralog was detected. Another the program is BaCoCa, which caclulates different alignment-based parameters for phylogenomic datasets.
As part of the FRIPRO project funded by the NFR called “InvertOmics – Phylogeny and evolution of lophotrochozoan invertebrates based on genomic data” to address these questions with respect to the phylogeny of Spiralia/Lophotrochozoa. In this project, besides aiming to generate high-quality genomes for 50 species covering all spiralian/lophotrochozoan phyla, we will also develop novel and innovative bioinformatic methods and tools. These will allow us to ameliorate the effects of the misleading biases even better. Moreover, this will also include a new support measurement, which is entirely different from all recent measurements. Due to both these new tools and genomes, we will be able find out how the different spiralian/lophotrochozoan phyla are related to each other and how the last common ancestor of animals like humans, insects, and earthworms looked and how evolution proceeded within this large group.
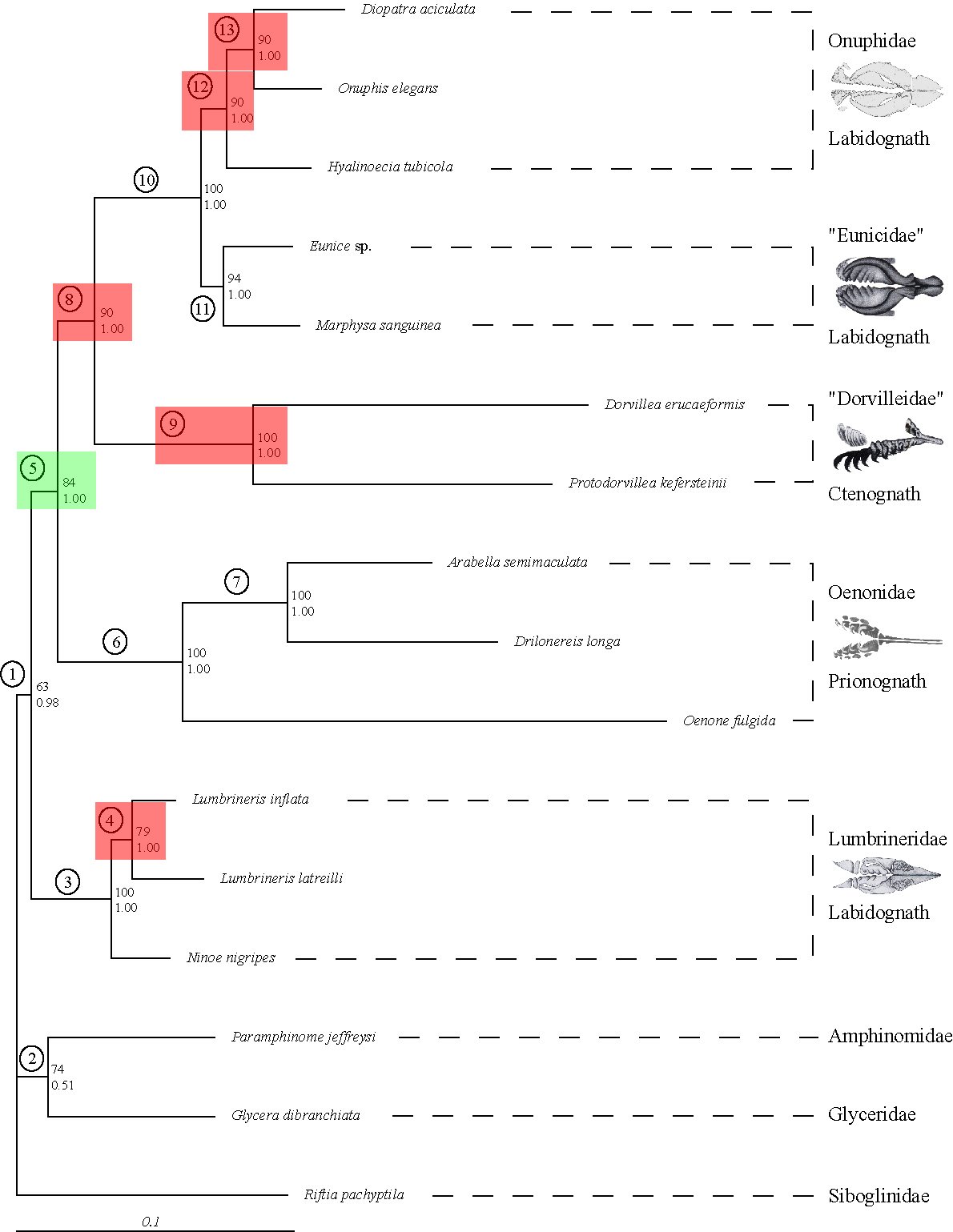
Previously, we brought some of these methods to bear on potential Long Branch problems such as might be the case with Platyzoa. The taxa grouping together as Platyzoa are characterized by strongly increased substitutions rates in molecular-phylogenetic or phylogenomic studies. However, such long-branched taxa have a tendency to artificially group together and, thus, it is uncertain if Platyzoa is a real monophyletic taxon with an overall increased substitution rate. Therefore, detection of such problems in crucial to reveal the phylogenetic signal regarding platyzoan taxa. In a recent study, we could convincingly show that indeed support for the monophyly of Platyzoa stemmed from artifical rather than real signal (see below).

Members of bioinformatic projects:
James Fleming
Stian Aleksander Helsem
Kevin M. Kocot (University of Alabama)
Nickellaus Roberts (University of Alabama)
John P. Huelsenbeck (University of Berkeley)
Previous projects:
- NSF-ATOL-10-513 “Collaborative Research: WormNet II: Assembling the Annelid Tree of Life”
- DFG-STR-683/7-1 “AnnEvol – Evolution and phylogeny of Annelida based on phylogenomic data (i.e. EST libraries)”
- DFG-STR-683/6-1 & 6-2 “Phylogenetic position of interstitial polychaete taxa of supposedly paedomorphic origin (i.e., Dinophilidae, Protodrilida, Polygordiidae and Nerillidae) using mitogenomic data”
- DFG-STR-683/5-2 “Phylogenomics of worm-like lophotrochozoans and platyzoans (Nemertea, Gastrotricha, Gnathostomulida)” within DFG priority program SPP 1174 “Deep Metazoan Phylogeny – Stammesgeschichte der Großgruppen der Tiere”
- DFG-Pu-84/5-1 “Phylogenomics of worm-like lophotrochozoans (Annelida, Sipuncula, Nemertea, Platyzoa)” within DFG priority program SPP 1174 “Deep Metazoan Phylogeny – Stammesgeschichte der Großgruppen der Tiere”
- DFG-STR-683/3-1 “Phylogenetic position of Annelida and possible subordinated annelid taxa (e.g. Sipuncula and Myzostomida) within Lophotrochozoa based on molecular data” within DFG priority program SPP 1174 “Deep Metazoan Phylogeny – Stammesgeschichte der Großgruppen der Tiere”
